Discover and quantify novel transcripts and splicing variants
INVIEW Transcriptome Discover is a complete RNA-Seq service that provides unique molecular insights into the transcriptome of nearly any organism.
The service can be used to profile all RNA species, including non-coding RNAs. Through the use of strand-specific libraries (Fig.1), the product can also help evaluate both sense and antisense transcripts. The 150 bp paired-end reads can be conveniently used for organisms with longer genes, such as plants. This product is applicable to any sample, as a well-defined reference sequence is not necessary.
Applications
- Elucidate cellular function through transcript quantification
- Analyse specific genomic elements and their biological significance
- Characterise differences in RNA splicing like alternative splicing
- Identify antisense transcripts
- Determine transcribed strand of non-coding RNAs
- Mark boundaries of closely situated or overlapping genes
Highlights INVIEW Transcriptome Discover
- Highly sensitive detection of new transcripts or splicing variants
- Structural information from strand-specific reads
- Applicable to nearly any organism of interest
Product Specifications
- Established protocols for generation of strand-specific libraries
- RNA-seq on leading sequencing platform
- Flexible sequencing starting with 30 M guaranteed read pairs
- Expert BioIT data analysis
- Comprehensive data report with results presented in different formats
- Applicable to samples from nearly any organism
- Availability of reference sequence is not crucial
- Optional services including RNA isolation and ribosomal RNA (rRNA) depletion
INVIEW Transcriptome Discover is an inclusive RNA-seq service that uses next-generation sequencing to provide the most comprehensive information on expression, levels, sequence, structure and strand orientation of RNA types. Strand-specific libraries can be sequenced with long and accurate read lengths to quantitatively evaluate common and rare transcripts, as well as to detect novel transcripts or alternative splicing events.
What is your transcriptome project aiming for? Are you seeking for unbiased information for basic or medical research? Or are you analysing metabolic pathways in order to improve production processes?
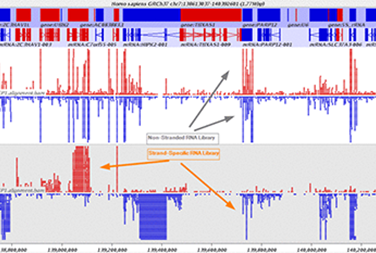
Our INVIEW Transcriptome packages provide standardized solutions that are suitable for a great variety of sequencing projects. Furthermore, raw data is processed within bioinformatics analysis and summarized in a comprehensive Data Analysis Report providing the results in a clear, understandable and informative way.
For projects without a reference genome, de novo transcriptome sequencing based on PacBio technology can be added to your project using our NGSelect product.
Learn more about the INVIEW Transcriptome Discover package
Starting Material
Accepted starting material for INVIEW Transcriptome Discover
- Total RNA from various sources
- rRNA depleted total RNA
Accepted starting material for RNA isolation
- Tissue
- Cells
- Body fluids
- FFPE tissue
Please note that only S1-classified material is accepted for RNA isolation ordered online. More information about the current classification of biological material can be found here (TRBA 466). Please contact us for further information on isolating RNA from material classified as S2.
Specifications
Optional pre-sequencing
- RNA isolation – efficient isolation of RNA for uniform and less-biased coverage
- Ribosomal RNA (rRNA) depletion – optimised removal of rRNA from mRNA for commonly studied organisms
- Please contact us to check the compatibility of your organism
Library types
- Standard eukaryotic library - mRNA is poly-(A) enriched and fragmented. cDNA synthesis is performed using random hexamer priming.
- Standard prokaryotic library - rRNA is removed by ribo depletion. cDNA synthesis is performed with random hexamer primers.
- 3’ fragment library – RNA is first fragmented, then the poly-(A)+ mRNA is enriched. In this library, each transcript is only represented by a short 3’ fragment, providing very deep and efficient expression analysis.
- Library with FFPE RNA - RNA isolation from Formalin Fixed Paraffin Embedded (FFPE) samples with optimised ribodepletion for reliable gene expression analysis and SNP detection.
- Strand-specific library
Sequencing platform
Sequencing mode
- 150 bp paired-end reads
- 30 million read pairs guaranteed
Bioinformatic analysis
- Semi-automatic mapping against one reference
- Identification of potential exon-exon splice junctions
- Identification and quantification of transcripts
- Merging of identified pieces to full length transcripts
- Transcript annotation based on known annotations
- Determination of expression levels
- Pairwise comparison of expression levels and determination of significant fold differences
- Detection and annotation of SNPs and InDels
- Annotation of detected SNPs and InDels that are registered in dbSNP
- Allocation of effects on protein level that are registered in Ensemble
Deliverables
- Alignment file (bam)
- Gene expression table including FPKM value (tsv)
- Combined gene expression table of all samples (tsv)
- Table of top genes expressed (tsv)
- Table of pair-wise differential gene expression including foldchange and p-value (tsv)
- Table of genes differently expressed between pairs (tsv)
- SNP and InDel tables including annotated variants and effects (vcf, tsv, bed)
- Comprehensive Data Analysis Report (.pdf)
Delivery time
- 21 days for up to 20 samples; every further 16 samples additional 7 days; 96 samples on request
- Express available: 17 days for up to 20 samples; every further 16 samples additional 6 days ; > 96 samples on request