1. How to Prepare Samples
Go to the sample submission guidelines for ONT/Whole Plasmid samples or Sanger samples for instructions on how to prepare your samples.
2. How to Label Samples
| 200 μL 96 well plate (>48 samples) - Write the order number along the side of the plate using a thin sharpie marker.
- After the order number, write ONT for Oxford Nanopore Samples (whole plasmid) or an S for Sanger samples.

- Make sure all handwriting is legible.
- Do not submit less than 48 samples per plate.
- Do not skip wells.
- Arrange samples by column (preferred.
| | 
|
3. How to Package Samples
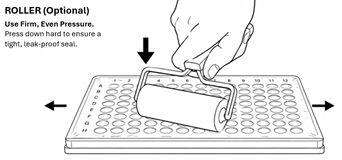
200 μL 96 well plate (>48 samples) - Heat seal is preferred. If using a roller on an adhesive seal, press down firmly and evenly across the entire plate to make sure it is securely sealed across all wells.
- It is critical that plates are sealed well to avoid leakage and cross contamination between wells.
- Inspect the seal for bubbles. Bubbles can cause cross contamination if liquid can pass between wells.
- Wrap plates individually with bubble wrap to protect them.
- Do not use cap strips on plates. These can cause cross contamination between wells when pulling them off.
- Avoid stacking multiple plates on top of each other. The wells from the top plate can piece the seal of the plate underneath.
Agar Plate - Wrap the agar plate in parafilm to prevent leakage and place inside a ziplock bag.
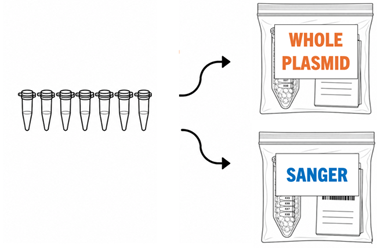
Separate Sample Types into Separate Bags - It is important to separate Whole Plasmid and Sanger samples into different ziplock bags so the processing team can quickly sort packages and send the samples to the right sequencing group.
- Include the order confirmation page in each bag, with the order barcode and sample information visible through the bag.
| | 
 |
4. How to Submit Samples
For more details, go to the How to Submit Samples webpage.
FAQ / Common Questions
Go to the FAQ page for all questions and answers regarding Whole Plasmid / Nanopore Sequencing.